|

|
Welcome to the ALOGPS 2.1 home page!
start the program
Java security issues: recently Java has dramatically increased security requirements to applets. Thus, please, follow instructions in this FAQ to correcly setup access to the software.
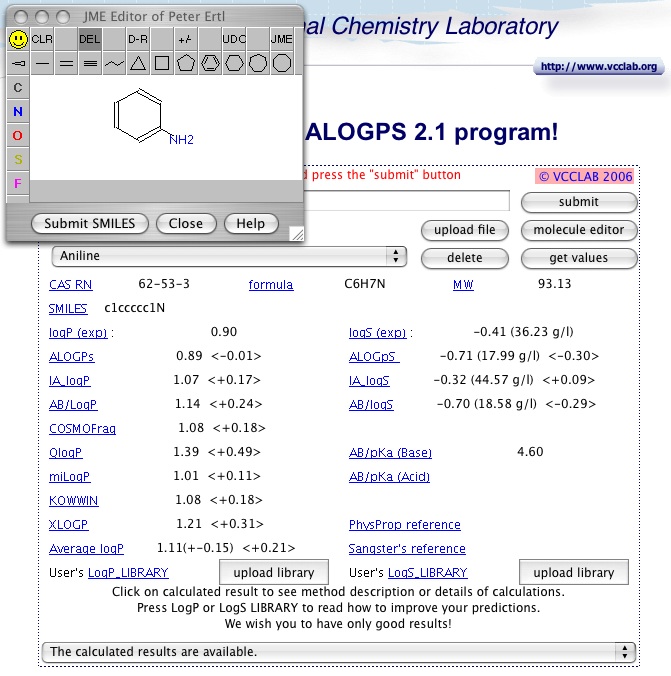
This Applet provides interactive on-line prediction of logP, water solubility and pKa(s) of compounds for drug design (ADME/T and HTS) and environmental chemistry studies.
In addition to the ALOGPS 2.1 logP and logW it also displays values calculated with
Pharma Algorithms LogP, LogS and pKa,
Actelion LogP & LogS (many thanks to Dr Thomas Sander),
Molinspiration logP,
KOWWIN logP,
ALOGP (Viswanadhan et al, 1989), MLOGP (Moriguchi et al, 1992) implemented in the DragonX software,
XLOGP2 and XLOGP3 programs and ChemAxon logP calculator.
The requests are sent to the corresponding servers and the results are
displayed in the applet.
Thus, you can compare several programs with one click. We thank all people who contributed their software!
If you do not have Java, have a look at typical output and
install it or use Non-Java Interface.
- 1-octanol/water calculation: ALOGPs was developed with 12908 molecules from the
PHYSPROP database using 75 E-state indicies. 64 neural networks were
trained using 50% of molecules selected by chance from the whole set.
The logP prediction accuracy is root mean squared error rms=0.35 and standard
mean error s=0.26 [1,2].
- Aqueous solubility calculation: ALOGpS was developed using 1291 molecules and provided
improved aqueous solubility prediction (rms=0.49, s=0.38) compared to our previous analysis [3]. The molecules used in this study can be downloaded.
ALOGPS 2.1 can increase its prediction
for the user's molecules up to 5 times [1] in the LIBRARY mode. You can create and use your
own LIBRARY. It can be also used to predict logD values [4,5].
Acknowledgment This software was developed with partial financial support from INTAS
and University of Lausanne.
References
- Tetko, I. V.; Tanchuk, V. Y. Application of associative neural networks for prediction of lipophilicity in ALOGPS 2.1 program, J. Chem. Inf. Comput. Sci., 2002, 42, 1136-45, article.
- Tetko, I. V.; Tanchuk, V. Y.; Villa, A. E. Prediction of n-octanol/water partition coefficients from PHYSPROP database using artificial neural networks and E-state indices, J. Chem. Inf. Comput. Sci., 2001, 41, 1407-21, article.
- Tetko, I. V.; Tanchuk, V. Y.; Kasheva, T. N.; Villa, A. E. Estimation of aqueous solubility of chemical compounds using E-state indices, J. Chem. Inf. Comput. Sci., 2001, 41, 1488-93, article.
- Tetko, I. V.; Poda, G. I. Application of ALOGPS 2.1 to predict log D distribution coefficient for Pfizer proprietary compounds, J. Med. Chem., 2004, 47, 5601-4, article.
- Tetko, I. V.; Bruneau, P. Application of ALOGPS to predict 1-octanol/water distribution coefficients, logP, and logD, of AstraZeneca in-house database, J Pharm Sci, 2004, 93, 3103-10, article.
- Poda, G. I.; Tetko, I. V.; Rohrer, D. C. Towards predictive ADME profiling of drug candidates: Lipophilicity and solubility, In 229th American Chemical Society National Meeting & Exposition; ACS: San Diego, CA, 2005, p MEDI 514 article.
- Viswanadhan, V.N.; Ghose, A.K.; Revankar, G.R. & Robins, R.K. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships. 4. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certain naturally occurring nucleoside antibiotics. J. Chem. Inf. Comput. Sci., 1989, 29, 163-172.
- Moriguchi, I.; Hirono, S.; Liu, Q.; Nakagome, I. & Matsushita, Y. Simple method of calculating octanol/water partition coefficient. Chem. Pharm. Bull., 1992, 40, 127-130.
 
|
|
|
|
{kind=link}